22. Example of ORA and GSEA#
We first run the same steps as in the previous notebook on multiple testing.
import pandas as pd
import numpy as np
from scipy.stats import ttest_ind
import sys
IN_COLAB = 'google.colab' in sys.modules
if IN_COLAB:
![ ! -f "dsbook/README.md" ] && git clone https://github.com/statisticalbiotechnology/dsbook.git
my_path = "dsbook/dsbook/common/"
else:
my_path = "../common/"
sys.path.append(my_path) # Read local modules for tcga access and qvalue calculations
import load_tcga as tcga
import qvalue
brca = tcga.get_expression_data(my_path + "../data/brca_tcga_pub2015.tar.gz", 'https://cbioportal-datahub.s3.amazonaws.com/brca_tcga_pub2015.tar.gz',"data_mrna_seq_v2_rsem.txt")
brca_clin = tcga.get_clinical_data(my_path + "../data/brca_tcga_pub2015.tar.gz", 'https://cbioportal-datahub.s3.amazonaws.com/brca_tcga_pub2015.tar.gz',"data_clinical_sample.txt")
brca.dropna(axis=0, how='any', inplace=True)
brca = brca.loc[~(brca<=0.0).any(axis=1)]
brca = pd.DataFrame(data=np.log2(brca),index=brca.index,columns=brca.columns)
brca_clin.loc["3N"]= (brca_clin.loc["PR_STATUS_BY_IHC"]=="Negative") & (brca_clin.loc["ER_STATUS_BY_IHC"]=="Negative") & (brca_clin.loc["IHC_HER2"]=="Negative")
triple_negative_bool = (brca_clin.loc["3N"] == True)
def get_significance_two_groups(row):
log_fold_change = row[triple_negative_bool].mean() - row[~triple_negative_bool].mean()
p = ttest_ind(row[triple_negative_bool],row[~triple_negative_bool],equal_var=False)[1]
return [p,-np.log10(p),log_fold_change]
pvalues = brca.apply(get_significance_two_groups,axis=1,result_type="expand")
pvalues.rename(columns = {list(pvalues)[0]: 'p', list(pvalues)[1]: '-log_p', list(pvalues)[2]: 'log_FC'}, inplace = True)
qvalues = qvalue.qvalues(pvalues)
File extracted to ../data/brca_tcga_pub2015
File extracted to ../data/brca_tcga_pub2015
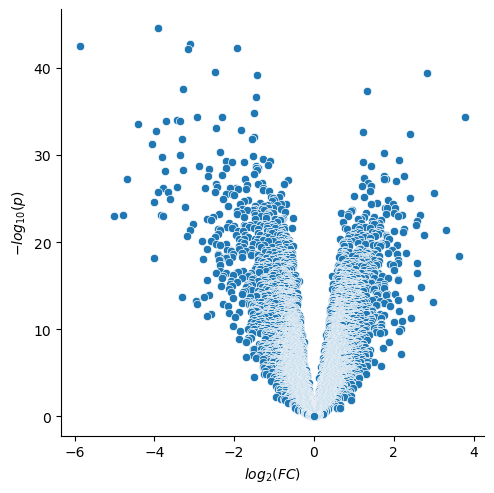
If we investigate a Volcano plot of the triple negative cancers vs. the other cancers, we see a large number of both up and down regulated genes. In this notebook we will examine if there are common patterns in the up and down regulation.
import matplotlib.pyplot as plt
import seaborn as sns
sns.relplot(data=qvalues,x="log_FC",y="-log_p")
plt.xlabel("$log_2(FC)$")
plt.ylabel("$-log_{10}(p)$")
plt.show()
22.1. Over-representation analysis#
We use the gseapy module to run an overrepresentation analysis. The module is unfortunately not implementing pathway analysis itself. It instead call a remote webserverEnrichr.
In the analysis here we use the KEGG database’s definition of metabolomic pathways. This choice can easily be changed to other databases such as GO.
Here we select to use the \(q\) values below \(10^{-12}\) as an input for the analysis. First we select this as our gene_list, and then we calculate the overlap of the gene list to all the pathways in KEGG.
import gseapy as gp
pathway_db=['KEGG_2019_Human']
background=set(qvalues.index)
gene_list = list(qvalues.loc[qvalues["q"]<1e-12,"q"].index)
output_enrichr=pd.DataFrame()
enr=gp.enrichr(
gene_list=gene_list,
gene_sets=pathway_db,
background=background,
outdir = None
)
len(gene_list)
1403
We clean up the results a bit by only keeping some of the resulting metrics. We also multiple hypothesis correct our results, and list the terms with a FDR less than 20%.
kegg_enr = enr.results[["P-value","Term"]].rename(columns={"P-value": "p"})
kegg_enr = qvalue.qvalues(kegg_enr)
kegg_enr.loc[kegg_enr["q"]<0.20]
| p | Term | q | |
|---|---|---|---|
| 0 | 0.000002 | Cell cycle | 0.000701 |
| 1 | 0.000265 | Hedgehog signaling pathway | 0.037638 |
| 2 | 0.001870 | Prostate cancer | 0.176826 |
The analysis seem to find overrepresentation of relatively few pathways, particularly given the significance of the differences between case and controll on transcript level.
22.2. Geneset Enrichment analysis#
Subsequently we us gseapy to perform a geneset enricment analysis (GSEA). This time we compare to pathways in GSEAs oncogenic signature database, MSigDB.
classes = ["tripleNeg" if triple_negative_bool[sample_name] else "Respond" for sample_name in brca.columns]
gs = gp.GSEA(data=brca,
gene_sets='MSigDB_Oncogenic_Signatures',
# gene_sets='KEGG_2019_Human',
classes=classes, # cls=class_vector
permutation_type='phenotype', # null from permutations of class labels
permutation_num=2000, # reduce number to speed up test
min_size=15, # minimal size of pathway
outdir=None, # do not write output to disk
no_plot=True, # Skip plotting
method='signal_to_noise',
threads=4, # Number of allowed parallel processes
seed=42,
format='png',)
# ascending=True)
gs.run()
gs_res = gs.res2d
2025-11-20 15:05:07,781 [WARNING] Found duplicated gene names, values averaged by gene names!
We first list the pathways with a FDR < 0.5.
significant = gs_res[gs_res["FDR q-val"]<0.5]
significant
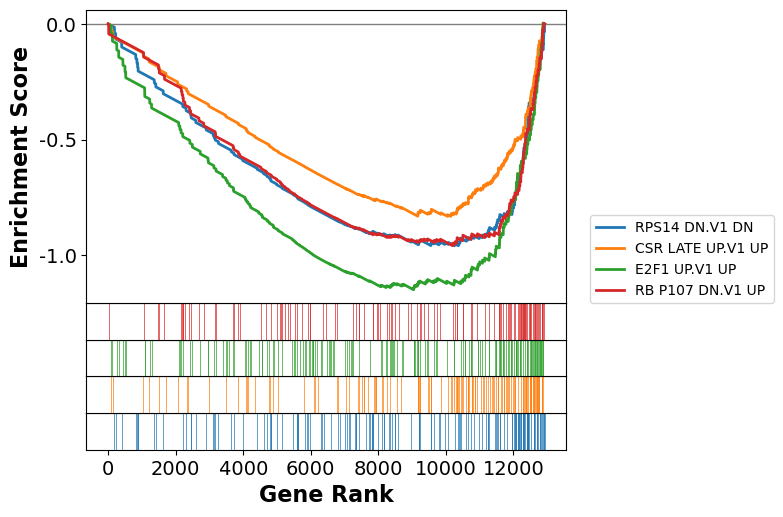
| Name | Term | ES | NES | NOM p-val | FDR q-val | FWER p-val | Tag % | Gene % | Lead_genes | |
|---|---|---|---|---|---|---|---|---|---|---|
| 0 | gsea | RPS14 DN.V1 DN | -0.506616 | -1.809224 | 0.018145 | 0.424336 | 0.1695 | 69/152 | 20.65% | PSAT1;CLCN4;SUV39H2;CDC20;MCM5;KIF2C;CDCA3;ASN... |
| 1 | gsea | CSR LATE UP.V1 UP | -0.572861 | -1.771695 | 0.014911 | 0.308419 | 0.2205 | 81/138 | 21.54% | LYAR;FANCA;CDCA8;MRAS;SNRPA1;FOXM1;CHEK2;RPF2;... |
| 3 | gsea | E2F1 UP.V1 UP | -0.448257 | -1.711008 | 0.01608 | 0.354616 | 0.318 | 82/167 | 30.03% | NDUFAF4;E2F3;AURKB;PIM1;KIFC1;CHEK1;UCK2;NASP;... |
| 5 | gsea | RB P107 DN.V1 UP | -0.518007 | -1.639151 | 0.052427 | 0.47894 | 0.4595 | 56/122 | 21.09% | CCNE1;STMN1;FANCA;MCM7;MCM5;ORC6;GTSE1;CDC25B;... |
The package we are using for accessing GSEA, gseapy, has some built in plotting routine for illustrating the enrichment for any given pathway.
axs = gs.plot(terms=significant.Term[0])

We can also compare the enrichment between multiple pathways
axs = gs.plot(significant.Term, show_ranking=False, legend_kws={'loc': (1.05, 0)}, )
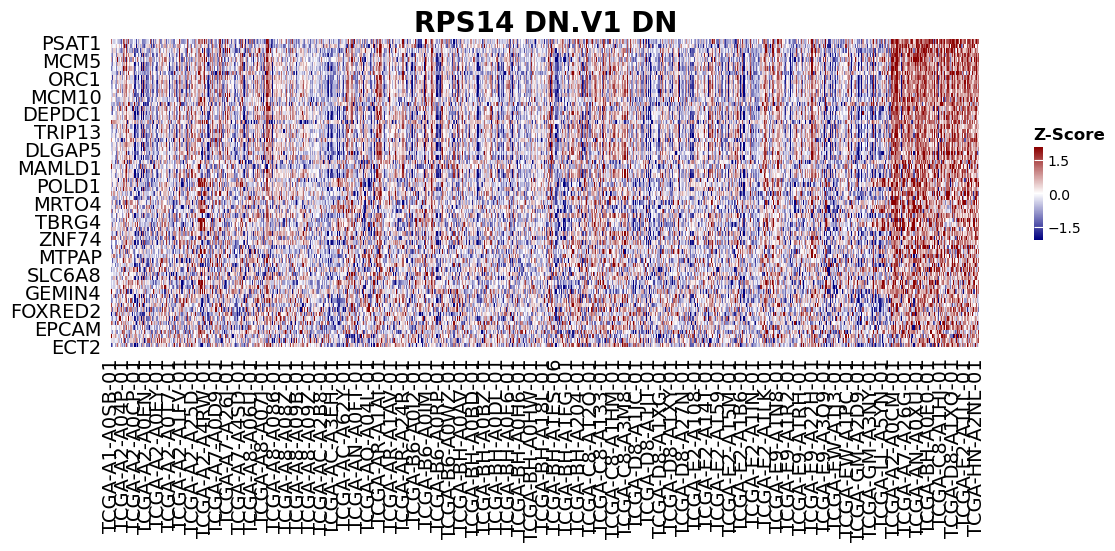
as well as the heatmap (Normalized gene expression as a function of Gene and patient) for the genes in a given pathway.
from gseapy import heatmap
# plotting heatmap
i = 0
genes = gs.res2d.Lead_genes[i].split(";")
# Make sure that ``ofname`` is not None, if you want to save your figure to disk
ax = heatmap(df = gs.heatmat.loc[genes], z_score=0, title=gs_res.Term[i], figsize=(14,4))